GuidelinesBy procedure2026.07.08
Medical Device Cybersecurity Guideline for Regulatory Review
Key cybersecurity review guidelines selected by CLARE Partners for preparing submission dossiers for connected medical devices in Korea.
Key takeaway — The moment a device has a communication function, cybersecurity is no longer optional — it is a review item. What is required is not a list of hacking concerns, but a documentation system flowing from risk management → security controls → configuration specification → post-market response plan, and the review scope covers not only "secure by design" but also post-market response.
For connected medical devices, cybersecurity is no longer a "security document" — it is a "regulatory strategy."
In medical devices, Wi-Fi, Bluetooth, USB, LAN, RS-232, app integration, server integration, and cloud storage are no longer add-on features. But the moment such a communication path is included, the regulatory review examines not only product performance but also cybersecurity risk management, security requirements, verification evidence, and the post-market response system.
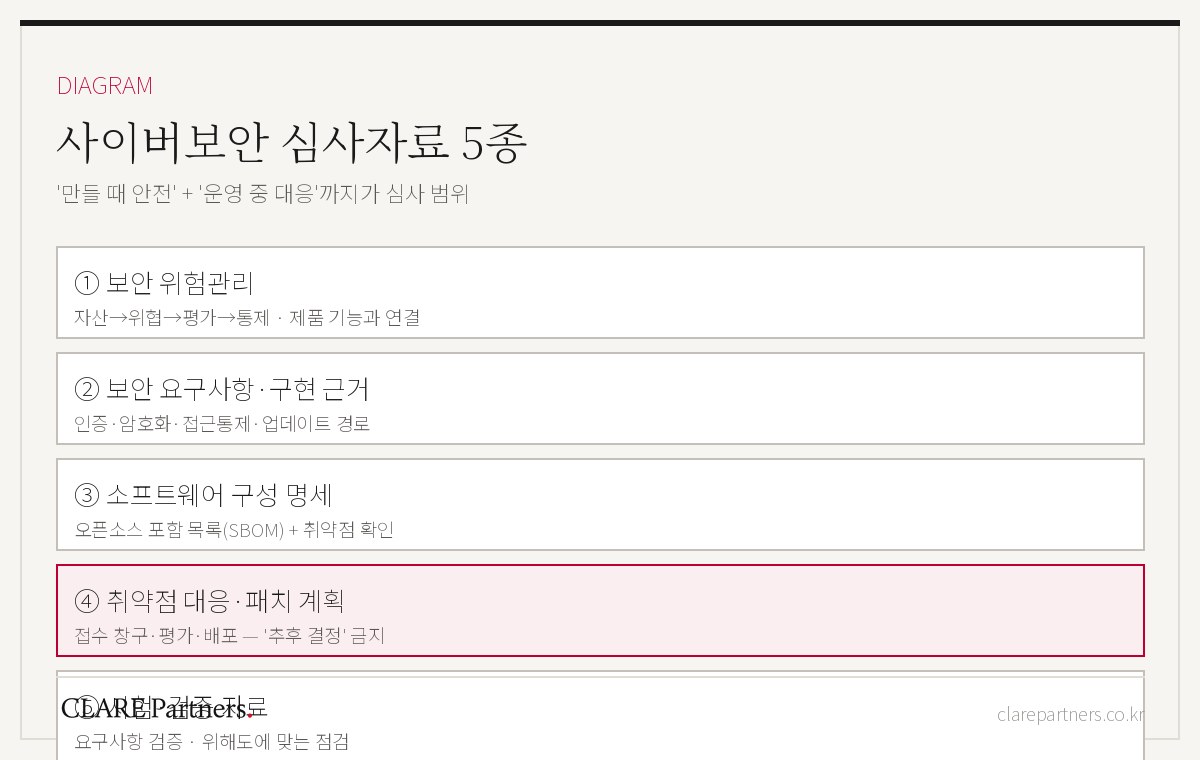
Through its Guideline on Cybersecurity Review and Approval of Medical Devices, the MFDS (Ministry of Food and Drug Safety) sets out cybersecurity requirements and the scope of submission documents for medical devices with wired or wireless communication paths, devices containing firmware or software, and software as a medical device (SaMD).
CLARE Partners treats cybersecurity documentation not as a mere attachment, but as a core review area for verifying consistency between the product architecture and the entire submission dossier.
CLARE Cybersecurity Readiness Framework
When reviewing cybersecurity documentation for a connected medical device, CLARE Partners first breaks the product down along four perspectives.
1. Interface
Check whether the product has any path connecting it to the outside.
Wi-Fi, Bluetooth, USB, LAN, RS-232, NFC, server communication, app integration, and hospital network integration can all fall within the scope of cybersecurity review. What matters in the review is not "whether the device connects to the internet," but whether any path exists that allows external access or data movement.
2. Data Flow
Check what data is generated, stored, and transmitted.
You need to map where patient information, measurements, diagnostic results, user account information, logs, settings, and update files move from and to. Until the data flows are mapped, the scope of encryption, access control, integrity verification, and audit logging cannot be defined clearly either.
3. User & Authority
Check who can access the product and with what privileges.
Access parties must be distinguished — users, administrators, clinicians, service engineers, hospital IT administrators, external servers, and mobile apps. Account management, authentication, authorization, session lock, and login-failure limits are all written against this structure.
4. Lifecycle
Check whether security can be maintained after the product is on the market.
Cybersecurity is not something verified only at the time of approval. The documentation must cover who receives reports when a vulnerability is discovered post-market, how it is assessed, and how patches or user notifications will be delivered.
Which products should be checked first?
For the following products, it is best to confirm early in approval preparation whether cybersecurity requirements apply.
App-connected personal measuring devices such as blood glucose meters, blood pressure monitors, and thermometers Equipment connected to hospital networks Remote monitoring medical devices Medical devices integrated with servers or the cloud Equipment capable of sending and receiving data via USB or LAN ports Medical devices with firmware update functions Software medical devices and SaMD Digital medical devices and in vitro diagnostic devices with communication functions
When judging whether a product is subject to cybersecurity review, do not look only at the product name or class — consider the communication paths, use environment, risk level, and data processing methods together.
What the review examines is not "the existence of security features."
The most common mistake in cybersecurity documentation is simply listing security features.
For example, statements like the following are not sufficient on their own.
"Login function available" "Encryption applied" "Access rights managed" "Update function available" "Security vulnerabilities to be addressed"
What the regulatory review requires is not whether a feature exists, but evidence answering the following questions.
Which interface does it apply to? Which users does it apply to? Which data does it apply to? Which risk is it intended to reduce? How was it implemented? How was it verified? How does it link to the risk management file?
In other words, the cybersecurity dossier is not an IT security manual — it is a regulatory document connected to the technical file, software documentation, and risk management file of the medical device.
Key deficiency points CLARE watches for
Cybersecurity deficiencies are often not resolved by fixing a single document. The product architecture, software versions, test data, and risk management file are all interconnected.
CLARE Partners focuses in particular on the following items.
Communication functions described in the technical file but missing from the cybersecurity documentation Accessible interfaces such as USB, LAN, Bluetooth, or Wi-Fi omitted from scope Cybersecurity risk analysis not linked to the risk management file Unclear separation of privileges among user, administrator, and service accounts Audit log items listed, but with unclear generation, storage, and retrieval methods An update function present, but no evidence of authenticity and integrity verification of update files No organized list of open-source or commercial software components No post-market procedure for receiving, assessing, and patching vulnerabilities Software version changes during development not reflected in the verification evidence
Because these items easily lead to deficiency (request for supplementation) during review, it is safer to manage them throughout development and verification rather than assembling them just before filing.
When should cybersecurity documentation be prepared?
The best time is not after product development is fully complete. It is most efficient to start once the communication architecture and software architecture are fixed.
At the design stage, map the communication paths, user types, data flows, and access privileges. At the verification stage, secure test or confirmation evidence for each security requirement. At the filing stage, align the technical file, software documentation, risk management file, and cybersecurity checklist for consistency.
In particular, if a product targets both Korean approval and overseas registrations, it is more efficient to design a common structure from the start than to build the Korean dossier first and rebuild overseas submissions later.
About the attachment
The attached document is the Guideline on Cybersecurity Review and Approval of Medical Devices published by the MFDS.
It is a useful reference for confirming the scope of application, cybersecurity requirements, range of submission documents, and checklists when preparing approvals for connected medical devices, software medical devices, digital medical devices, and in vitro diagnostic devices.
CLARE Partners Comment
Cybersecurity is not a standalone document appended at the end of the dossier. It is an area where the product's communication architecture, data flows, user privileges, software verification, and risk management file must align as one coherent whole.
CLARE Partners reviews the cybersecurity scope of each product from a medical device regulatory perspective and designs a submission package that is consistent with the technical file, software documentation, and risk management file.
If you are preparing a connected medical device or SaMD product, we recommend scoping your cybersecurity documentation before filing for approval.
Frequently asked questions
- Q. Which products are subject to cybersecurity review documentation?
- Medical devices with wired or wireless communication functions. Typical examples include app-connected measuring devices, hospital equipment connected to a network, remote monitoring devices, and software as a medical device (SaMD). Even if the communication module is "optional," it falls within scope as long as it is installed.
- Q. Does a simple device that only connects to an app via Bluetooth also fall within scope?
- Yes. The mere existence of a communication path is the starting point of the risk analysis. However, the depth of documentation required is proportional to the device's risk level and to what the communication actually does (simple data viewing versus changing therapy settings).
- Q. Do we need to prepare separate cybersecurity documents for Korea and for the FDA?
- The required frameworks of the two systems (risk management, security controls, configuration specification, and vulnerability response) overlap to a large degree. In our experience, designing both frameworks together from the start makes extending the Korean dossier into the FDA structure easier than the reverse.