ガイドライン手続き別2026.07.08
医療機器サイバーセキュリティ承認審査ガイドライン
CLARE Partnersが通信機能付き医療機器の承認資料準備のために厳選した、サイバーセキュリティ承認・審査の要点ガイドラインです。
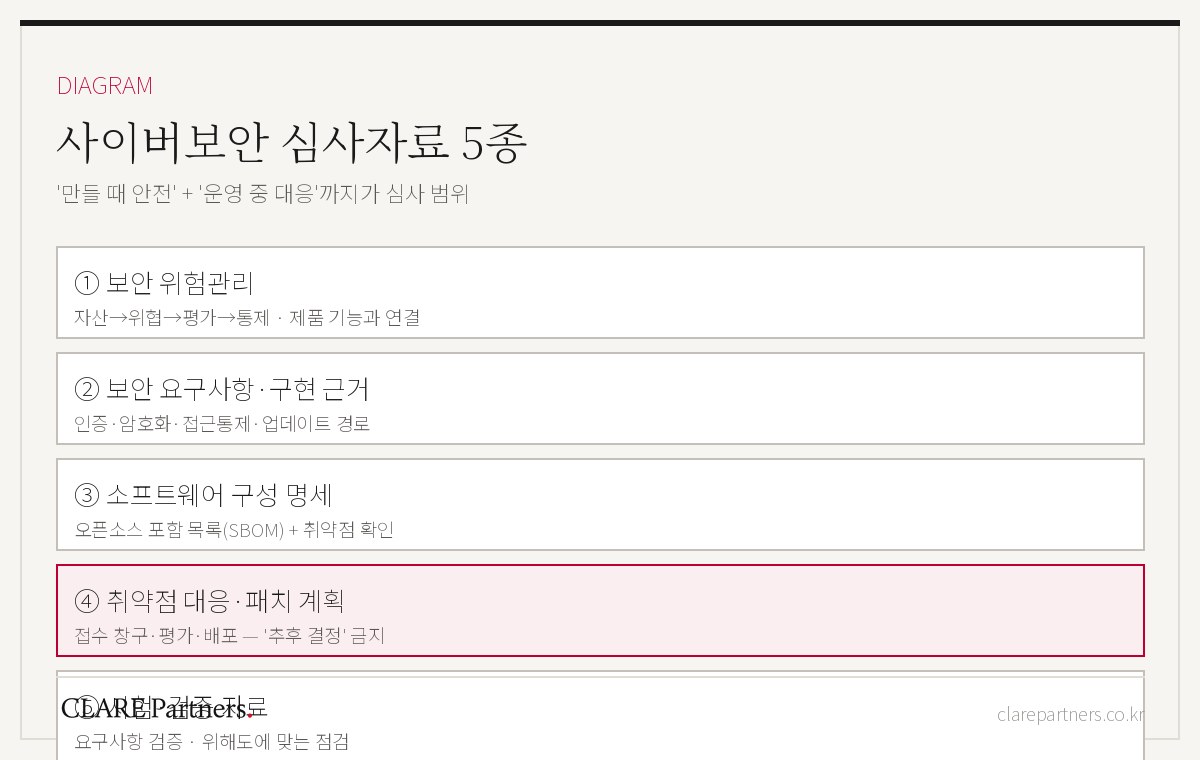
要点サマリー — 通信機能がある時点で、サイバーセキュリティは選択肢ではなく審査項目です。求められるのはハッキングへの不安の羅列ではなく、リスクマネジメント → セキュリティコントロール → 構成明細 → 運用中の対応計画へとつながる文書体系であり、「作る時の安全」だけでなく「市販後の対応」までが審査範囲です。
通信機能のある医療機器 — もはや「セキュリティ資料」ではなく「承認戦略」です
医療機器において、Wi-Fi、Bluetooth、USB、LAN、RS-232、アプリ連携、サーバー連携、クラウド保存機能は、もはや付加機能ではありません。 しかし、こうした通信経路が含まれた瞬間、承認・審査の段階では製品の性能だけでなく、サイバーセキュリティリスクマネジメント、セキュリティ要求事項、検証資料、市販後対応体制まで併せて審査されます。
韓国食品医薬品安全処(MFDS)は「医療機器のサイバーセキュリティ承認・審査ガイドライン」を通じて、有線・無線の通信経路を持つ医療機器、ファームウェアまたはソフトウェアを含む医療機器、ソフトウェア医療機器(SaMD)に対し、サイバーセキュリティ要求事項と提出資料の範囲を提示しています。
CLARE Partnersは、サイバーセキュリティ資料を単なる添付文書ではなく、製品構造と承認資料全体の整合性を確認する中核的な検討領域と捉えています。
CLARE Cybersecurity Readiness Framework
CLARE Partnersは、通信機能を持つ医療機器のサイバーセキュリティ資料を検討する際、まず次の4つの観点から製品を分解します。
1. Interface
製品に外部とつながる経路があるかを確認します。
Wi-Fi、Bluetooth、USB、LAN、RS-232、NFC、サーバー通信、アプリ連携、病院ネットワーク連携などは、いずれもサイバーセキュリティ検討の対象になり得ます。 審査で重要なのは「インターネットに接続されているか」ではなく、外部からのアクセスまたはデータ移動が可能な経路が存在するかです。
2. Data Flow
どのようなデータが生成され、保存され、送信されるかを確認します。
患者情報、測定値、診断結果、ユーザーアカウント情報、ログ、設定値、アップデートファイルなどが、どこからどこへ移動するのかを整理する必要があります。 データフローが整理されていなければ、暗号化、アクセスコントロール、完全性検証、監査ログの適用範囲も明確になりません。
3. User & Authority
誰が製品にアクセスでき、どのような権限を持つかを確認します。
ユーザー、管理者、医療従事者、サービスエンジニア、病院のIT管理者、外部サーバー、モバイルアプリなど、アクセス主体を区別する必要があります。 アカウント管理、認証、権限付与、セッションロック、ログイン失敗回数制限などは、この構造を基準に作成されます。
4. Lifecycle
製品の発売後もセキュリティが維持できるかを確認します。
サイバーセキュリティは承認時点だけで確認する資料ではありません。 市販後に脆弱性が発見された際、誰が受け付け、どのように評価し、どのような方法でパッチを提供し、あるいはユーザーに案内するのかまで文書化される必要があります。
どのような製品から先に点検すべきでしょうか
次の製品は、承認準備の初期段階でサイバーセキュリティの適用有無を先に確認することをおすすめします。
アプリ連動型の血糖測定器、血圧計、体温計などの個人用測定機器 病院ネットワークに接続される装置 遠隔モニタリング医療機器 サーバーまたはクラウドと連携する医療機器 USBまたはLANポートを通じてデータの送受信が可能な装置 ファームウェアアップデート機能を持つ医療機器 ソフトウェア医療機器、SaMD デジタル医療機器および体外診断用医療機器のうち通信機能を持つ製品
サイバーセキュリティ検討対象かどうかを判断する際は、製品名やクラスだけを見るのではなく、通信経路、使用環境、リスクの大きさ、データ処理方式を併せて見る必要があります。
審査で見られる核心は「セキュリティ機能の存在」ではありません
サイバーセキュリティ資料で最もよくある間違いは、セキュリティ機能を単に羅列することです。
たとえば、次のような表現だけでは十分ではありません。
「ログイン機能あり」 「暗号化を適用」 「アクセス権限管理」 「アップデート機能あり」 「セキュリティ脆弱性への対応予定」
承認・審査で必要なのは機能の有無ではなく、次の問いに対する根拠です。
どのインターフェースに適用されるのか どのユーザーに適用されるのか どのデータに適用されるのか どのリスクを低減するための機能なのか どのように実装されたのか どのように検証されたのか リスクマネジメント文書とどのようにつながるのか
つまり、サイバーセキュリティ資料はITセキュリティの説明書ではなく、医療機器の技術文書、ソフトウェア資料、リスクマネジメント文書とつながる承認資料なのです。
CLAREが注目する主な補完(追加資料要求)ポイント
サイバーセキュリティ資料は、文書を1つ修正するだけでは解決しないケースが多くあります。 製品構造、ソフトウェアバージョン、試験資料、リスクマネジメント文書が相互に結びついているためです。
CLARE Partnersは特に次の項目を重点的に確認します。
通信機能が技術文書には記載されているが、サイバーセキュリティ資料からは漏れているケース USB、LAN、Bluetooth、Wi-Fiなどアクセス可能なインターフェースが抜けているケース リスクマネジメント文書とサイバーセキュリティリスク分析がつながっていないケース ユーザー、管理者、サービスアカウントの権限区分が不明確なケース 監査ログの項目はあるが、生成・保存・照会の方式が不明確なケース アップデート機能はあるが、アップデートファイルの真正性・完全性検証の根拠がないケース オープンソースまたは商用ソフトウェア構成要素のリストが整理されていないケース 市販後の脆弱性の受付、評価、パッチ配布の手順がないケース 開発中に変更されたソフトウェアバージョンが検証資料に反映されていないケース
これらの項目は審査段階で補完(追加資料要求)につながりやすいため、承認申請の直前に整理するのではなく、開発・検証段階から併せて管理するのが安全です。
サイバーセキュリティ資料はいつ準備すべきでしょうか
最適なタイミングは、製品開発がすべて終わった後ではありません。 通信構造とソフトウェア構造が確定する時点から準備するのが最も効率的です。
設計段階では、通信経路、ユーザー類型、データフロー、アクセス権限を整理する必要があります。 検証段階では、セキュリティ要求事項ごとの試験または確認資料を確保する必要があります。 承認準備段階では、技術文書、ソフトウェア資料、リスクマネジメント文書、サイバーセキュリティチェックリストの整合性を合わせる必要があります。
特に韓国での承認と海外での許認可を併せて視野に入れる製品であれば、韓国向け資料を先に作って後から海外提出資料を作り直す方式より、最初から共通の構造を設計する方が効率的です。
添付ファイルのご案内
添付資料は、韓国食品医薬品安全処(MFDS)が発行した「医療機器のサイバーセキュリティ承認・審査ガイドライン」です。
通信機能を持つ医療機器、ソフトウェア医療機器、デジタル医療機器、体外診断用医療機器の承認・審査準備の際に、適用対象、サイバーセキュリティ要求事項、提出資料の範囲、チェックリストを確認する参考資料としてご活用いただけます。
CLARE Partners Comment
サイバーセキュリティは、承認資料の最後に付け足す別文書ではありません。 製品の通信構造、データフロー、ユーザー権限、ソフトウェア検証、リスクマネジメント文書が一つの流れとして噛み合わなければならない領域です。
CLARE Partnersは、医療機器許認可の観点から製品ごとのサイバーセキュリティ適用範囲を検討し、技術文書・ソフトウェア資料・リスクマネジメント文書と整合性のある提出資料の構成を共に設計します。
通信機能を含む医療機器またはSaMD製品を準備中であれば、承認申請の前にサイバーセキュリティ資料の範囲をまず点検されることをおすすめします。
よくあるご質問
- Q. どのような製品がサイバーセキュリティ審査資料の対象になりますか?
- 有線・無線の通信機能を備えた医療機器が対象です。アプリと連動する測定機器、ネットワークに接続される病院設備、遠隔モニタリング機器、ソフトウェア医療機器(SaMD)が代表例です。通信モジュールが「オプション」であっても、搭載されていれば検討対象になります。
- Q. Bluetoothでアプリと接続するだけの単純な機器も該当しますか?
- はい。通信経路が存在するという事実自体がリスク分析の出発点になります。ただし、要求される文書の深さは、機器のリスクの大きさと通信が担う機能(単なる参照なのか、治療設定の変更なのか)に比例します。
- Q. 韓国向けとFDA向けのサイバーセキュリティ文書は別々に作成する必要がありますか?
- 両制度が要求する骨格(リスクマネジメント・セキュリティコントロール・構成明細・脆弱性対応)はかなりの部分で重なります。実務経験上、最初から両制度を重ねて設計すれば、韓国向け資料をFDAの構造に拡張する経路の方が、その逆よりも容易です。