指南按程序2026.07.08
医疗器械网络安全许可审评指南
CLARE Partners为具备通信功能的医疗器械整理的网络安全许可·审评核心指南,帮助企业准备许可资料。
核心摘要 — 一旦产品具备通信功能,网络安全就不再是可选项,而是审评项目。审评要求的不是罗列对黑客攻击的担忧,而是一套由风险管理 → 安全控制 → 配置清单 → 运营期应对计划串联起来的文件体系;审评范围不仅涵盖"设计时的安全",还包括"上市后的应对"。
具备通信功能的医疗器械,需要的不再是"安全资料",而是"许可战略"。
在医疗器械中,Wi-Fi、蓝牙、USB、LAN、RS-232、App联动、服务器联动、云端存储等功能已不再是附加功能。 然而,一旦产品包含这些通信路径,在许可·审评阶段,除了产品性能之外,网络安全风险管理、安全要求、验证资料、上市后应对体系也会被一并审查。
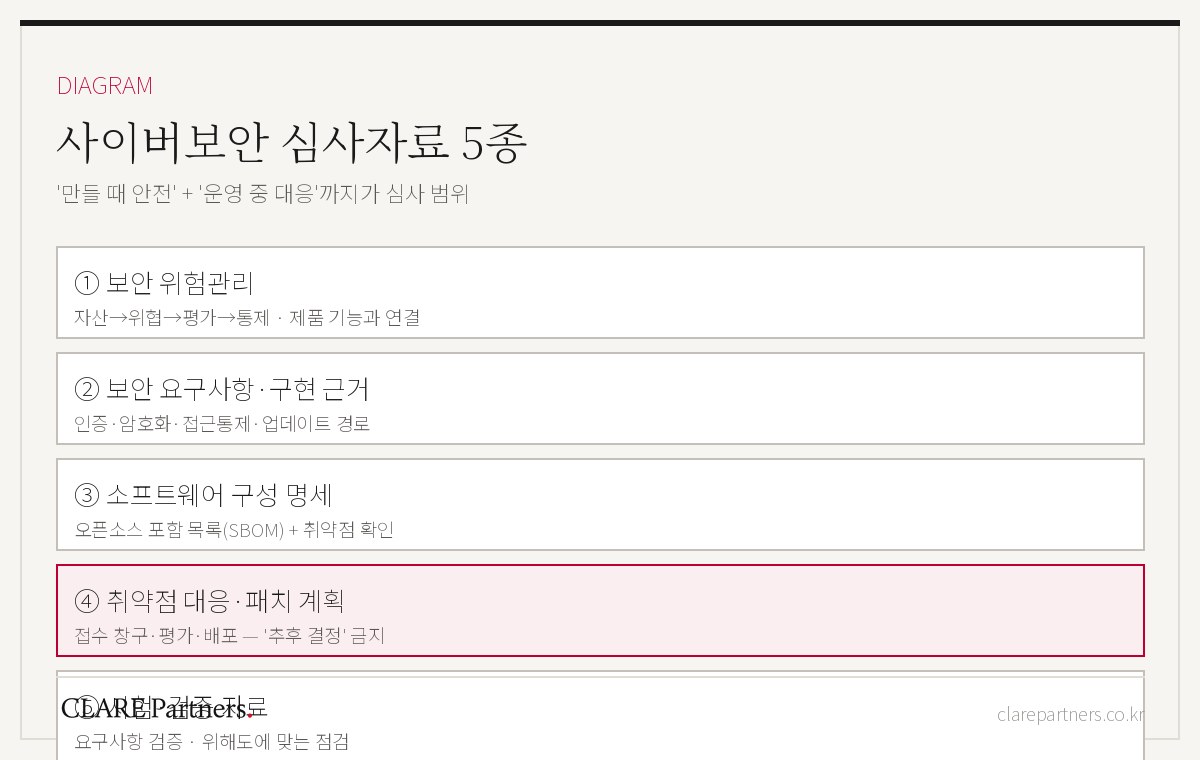
韩国食药处(MFDS)通过《医疗器械网络安全许可·审评指南》,针对具备有线·无线通信路径的医疗器械、包含固件或软件的医疗器械以及软件医疗器械(SaMD),提出了网络安全要求和提交资料的范围。
CLARE Partners并不将网络安全资料视为单纯的附件,而是将其定位为验证产品结构与整套许可资料一致性的核心审查领域。
CLARE Cybersecurity Readiness Framework
CLARE Partners在审查具备通信功能的医疗器械的网络安全资料时,会首先从以下四个视角对产品进行拆解。
1. Interface(接口)
确认产品是否存在与外部连接的路径。
Wi-Fi、蓝牙、USB、LAN、RS-232、NFC、服务器通信、App联动、医院网络联动等,都可能成为网络安全审查的对象。 审评中重要的不是"是否连接互联网",而是是否存在可供外部访问或数据流动的路径。
2. Data Flow(数据流)
确认哪些数据被生成、存储和传输。
需要梳理患者信息、测量值、诊断结果、用户账户信息、日志、设置值、更新文件等数据从何处流向何处。 如果数据流未被梳理清楚,加密、访问控制、完整性验证、审计日志的适用范围也就无从明确。
3. User & Authority(用户与权限)
确认谁可以访问产品,以及各自拥有何种权限。
需要区分用户、管理员、医护人员、维修工程师、医院IT管理员、外部服务器、移动App等访问主体。 账户管理、身份认证、权限授予、会话锁定、登录失败次数限制等内容,均以这一结构为基准编写。
4. Lifecycle(生命周期)
确认产品上市后安全性能否持续维持。
网络安全不是仅在许可时点确认一次的资料。 上市后一旦发现漏洞,由谁受理、如何评估、以何种方式发布补丁或告知用户,这些都必须形成文件。
哪些产品应当优先自查?
以下产品建议在许可准备的早期阶段,优先确认是否适用网络安全要求。
与App联动的血糖仪、血压计、体温计等个人用测量设备 连接医院网络的设备 远程监护医疗器械 与服务器或云端联动的医疗器械 可通过USB或LAN端口收发数据的设备 具备固件更新功能的医疗器械 软件医疗器械(SaMD) 数字医疗器械及体外诊断医疗器械中具备通信功能的产品
判断产品是否属于网络安全审查对象时,不能只看产品名称或类别等级,还必须综合考虑通信路径、使用环境、风险程度、数据处理方式。
审评关注的核心并非"安全功能是否存在"。
网络安全资料中最常见的失误,就是单纯罗列安全功能。
例如,仅有以下表述是不够的。
"具有登录功能" "已应用加密" "进行访问权限管理" "具有更新功能" "计划应对安全漏洞"
许可·审评所需要的不是功能是否存在,而是对以下问题的依据说明。
适用于哪个接口 适用于哪类用户 适用于哪些数据 用于降低何种风险 如何实现 如何验证 与风险管理文件如何关联
也就是说,网络安全资料不是IT安全说明书,而是与医疗器械技术文件、软件资料、风险管理文件相互关联的许可资料。
CLARE关注的主要补正风险点
网络安全资料的问题往往无法通过修改单份文件解决。 因为产品结构、软件版本、试验资料、风险管理文件是相互关联的。
CLARE Partners会重点审查以下事项。
技术文件中记载了通信功能,但网络安全资料中却遗漏的情况 USB、LAN、蓝牙、Wi-Fi等可访问接口被遗漏的情况 风险管理文件与网络安全风险分析未建立关联的情况 用户、管理员、维修账户的权限划分不明确的情况 虽列出审计日志项目,但其生成·存储·查询方式不明确的情况 虽具备更新功能,但缺乏更新文件真实性·完整性验证依据的情况 开源或商用软件组件清单未经整理的情况 缺少上市后漏洞受理、评估、补丁发布流程的情况 开发过程中变更的软件版本未反映在验证资料中的情况
上述事项在审评阶段极易导致补正,因此与其在提交许可申请前夕才匆忙整理,不如在开发·验证阶段就同步管理,才更为稳妥。
网络安全资料应在何时开始准备?
最佳时点并不是产品开发全部完成之后。 从通信结构和软件结构确定的时点开始准备,才是最高效的。
在设计阶段,应梳理通信路径、用户类型、数据流和访问权限。 在验证阶段,应针对各项安全要求取得试验或确认资料。 在许可准备阶段,应确保技术文件、软件资料、风险管理文件与网络安全检查表之间的一致性。
尤其是同时考虑韩国许可与海外注册的产品,与其先做韩国资料、之后再重做海外提交资料,不如从一开始就设计共通结构,效率更高。
附件说明
所附资料为韩国食药处(MFDS)发布的《医疗器械网络安全许可·审评指南》。
在准备具备通信功能的医疗器械、软件医疗器械、数字医疗器械、体外诊断医疗器械的许可·审评时,可参考其中的适用对象、网络安全要求、提交资料范围及检查表。
CLARE Partners Comment
网络安全不是附在许可资料末尾的独立文件。 它是一个需要产品的通信结构、数据流、用户权限、软件验证、风险管理文件形成统一逻辑的领域。
CLARE Partners从医疗器械注册的视角出发,审查各产品的网络安全适用范围,并协助企业构建与技术文件·软件资料·风险管理文件保持一致性的提交资料体系。
如果您正在准备包含通信功能的医疗器械或SaMD产品,建议在提交许可申请前,先行确认网络安全资料的范围。
常见问题
- Q. 哪些产品需要提交网络安全审评资料?
- 具备有线或无线通信功能的医疗器械均属于适用对象。典型例子包括与App联动的测量设备、连接医院网络的设备、远程监护设备以及软件医疗器械(SaMD)。即使通信模块只是"选配",只要实际搭载,就属于审查范围。
- Q. 仅通过蓝牙与App连接的简单设备也适用吗?
- 是的,存在通信路径这一事实本身就是风险分析的起点。不过,所要求文件的深度与器械的风险程度以及通信所执行的功能(是单纯查询数据,还是更改治疗设置)成正比。
- Q. 韩国国内用与FDA用的网络安全文件需要分别编制吗?
- 两套体系的核心框架(风险管理·安全控制·配置清单·漏洞应对)在很大程度上是重叠的。根据实务经验,从一开始就将两套体系叠加设计,把韩国资料扩展为FDA结构的路径,比反向操作更为顺畅。