가이드라인GMP 가이드2026.07.08
KGMP 적합인정 가이드 — 심사 종류, 준비 서류, 해외 제조소 대응
2등급 이상 의료기기의 통과의례, KGMP. 심사 종류별 차이, 품질시스템 문서의 뼈대, 해외 제조소 심사에서 프로젝트가 지연되는 진짜 이유를 정리했습니다.
핵심 요약 — KGMP는 "제품"이 아니라 **"만드는 시스템"**에 대한 심사입니다. 2등급 이상 의료기기의 사실상 통과의례이며, 수입 프로젝트에서는 해외 제조소의 협조 수준이 곧 일정입니다. ISO 13485가 있으면 유리하지만, 면제는 아닙니다.
KGMP가 보는 것
품목 인허가가 "이 제품이 안전·유효한가"를 본다면, KGMP(의료기기 제조 및 품질관리 기준)는 **"이 제조소가 그런 제품을 일관되게 만들 수 있는가"**를 봅니다. 심사 대상은 제조소의 품질시스템 — 문서, 기록, 설비, 인력, 그리고 그것들이 실제로 돌아가는 현장입니다.
2등급 이상 품목의 제조·수입에는 해당 제조소의 적합인정이 사실상 세트이며(등급별 절차 참고), 1등급도 멸균 등 일부 품목은 예외적으로 대상이 됩니다.
심사의 종류 — 언제 어떤 심사를 받나
| 심사 | 언제 | 특징 |
|---|---|---|
| 최초심사 | 그 제조소에서 처음 적합인정을 받을 때 | 문서+현장, 준비 부담 최대 |
| 추가심사 | 기존 적합 제조소에 품목군 추가 등 | 변경 범위 중심 |
| 변경심사 | 소재지·중요 공정 등 변경 시 | 변경 사항의 영향 평가 |
| 정기심사 | 유효기간 도래 시 갱신 | 시스템 유지 여부 확인 |
수입이라면 이 심사가 해외 제조소를 대상으로 이뤄지며, 자료 심사와 현장 심사의 조합은 사안에 따라 정해집니다.
품질시스템 문서의 뼈대
- 품질매뉴얼과 절차서 — 설계·구매·생산·검사·출하·불만처리·시정예방(CAPA)까지의 체계
- 제품표준서 계열 — 해당 품목을 '어떻게 만들고 어떻게 검사하는가'의 표준
- 기록 — 절차서대로 실제 운영했다는 증거. 심사에서 가장 많이 들춰지는 것은 매뉴얼이 아니라 기록입니다
- 밸리데이션 — 멸균, 특수 공정 등 결과로 검증 안 되는 공정의 사전 검증
- 교육·설비 관리 — 인력 자격과 설비 적격성
ISO 13485 체계가 있는 제조소라면 이 뼈대의 상당 부분이 이미 있습니다. 실무는 "있는 것"과 "한국이 요구하는 것"의 갭 분석에서 시작합니다.
해외 제조소 심사 — 지연의 진짜 이유
수입 프로젝트에서 KGMP가 늦어지는 원인은 대부분 기술이 아니라 협조 구조입니다.
"왜 우리가?"의 벽. 제조원 입장에서 KGMP는 남의 나라 심사입니다. 우선순위에서 밀리기 일쑤라, 계약서의 심사 협조 의무 조항이 유일하게 확실한 지렛대입니다.
문서 언어와 형식. 제조원 문서를 한국 심사가 읽을 수 있는 형태로 정리하는 작업량을 과소평가하면 일정이 무너집니다.
현장 준비. 심사 당일의 질문은 문서가 아니라 현장 기록으로 향합니다. 모의 점검 없이 현장 심사를 맞으면, 사소한 기록 공백이 보완으로 이어집니다.
준비 체크리스트
- 대상 제조소·품목군 확정, 심사 종류 확인
- 제조원 ISO 13485 등 기존 체계 확인 → 갭 분석
- 계약서에 자료 제공·현장 심사 협조 의무 반영
- 문서 취합·번역·정리 일정 확보
- 현장 모의 점검 계획
- 기술문서 심사와의 병행 일정 설계
KGMP는 제조원과의 협업 설계가 절반입니다. 제조소 정보(국가, 기존 인증, 품목군)를 무료 사전 검토로 보내주시면, 심사 종류와 준비 범위의 갭 분석 초안을 잡아 드립니다.
자주 묻는 질문
- Q. KGMP는 언제까지 받아야 하나요?
- 품목 인증·허가와 맞물려 있어, 제품을 출하·판매하려면 해당 제조소의 적합인정이 갖춰져 있어야 합니다. 기술문서 심사와 병행 준비하는 것이 전체 기간을 줄이는 정석입니다.
- Q. 해외 제조사가 ISO 13485 인증이 있으면 KGMP는 면제되나요?
- 면제되지 않습니다. ISO 13485는 KGMP 준비의 훌륭한 기반이지만, KGMP는 한국 요구사항에 따른 별도의 적합인정 절차입니다. 다만 13485 체계가 잘 갖춰진 제조소는 문서 갭 분석과 보완 범위가 크게 줄어듭니다.
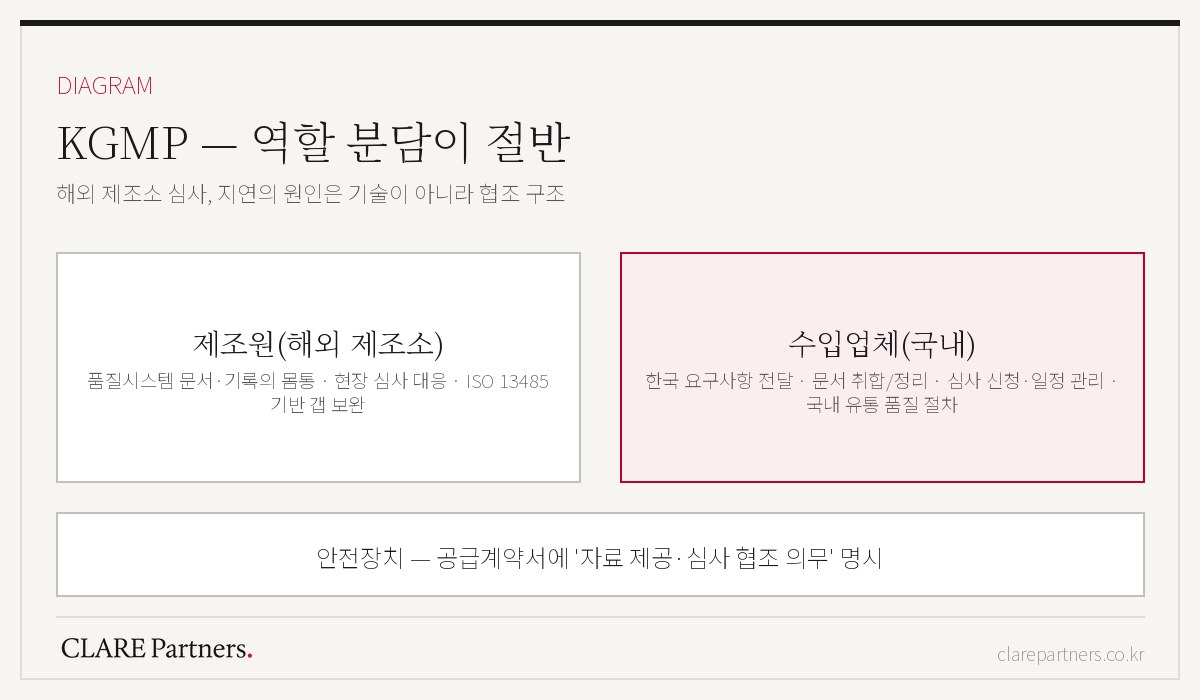
- Q. 수입업체인 우리가 준비할 것과 제조원이 준비할 것은 어떻게 나뉘나요?
- 품질시스템 문서와 현장 심사 대응의 몸통은 제조원 몫이고, 수입업체는 국내 요구사항 전달·문서 취합·심사 신청과 일정 관리, 그리고 국내 유통 단계의 품질 절차를 맡습니다. 이 역할 분담을 계약서에 명시하지 않으면 중간에서 프로젝트가 섭니다.