指南按类别2026.07.08
2类医疗器械认证指南
2类的正式程序名称是'认证'。这是技术文件审评、检测检验和KGMP首次登场的类别 — 本文整理了5个步骤流程以及左右周期的3大变量。
核心摘要 — 2类是韩国市场流通医疗器械中占比最大的类别,也是技术文件审评和KGMP正式登场的第一个类别。总费用和周期取决于"检测什么、可以省略什么"。
2类的正式名称是'认证'
2类是潜在风险较低的医疗器械。在程序上,属于由韩国食药处(MFDS)指定的认证机构进行技术文件审评的'认证'对象(新研发等部分情形属于许可对象),位于1类备案与3·4类许可之间。
从实务感觉来讲 — 如果说1类是"整理文件",那么从2类开始就进入了"举证"的领域。必须用检测数据和文件证明产品安全且具备性能。
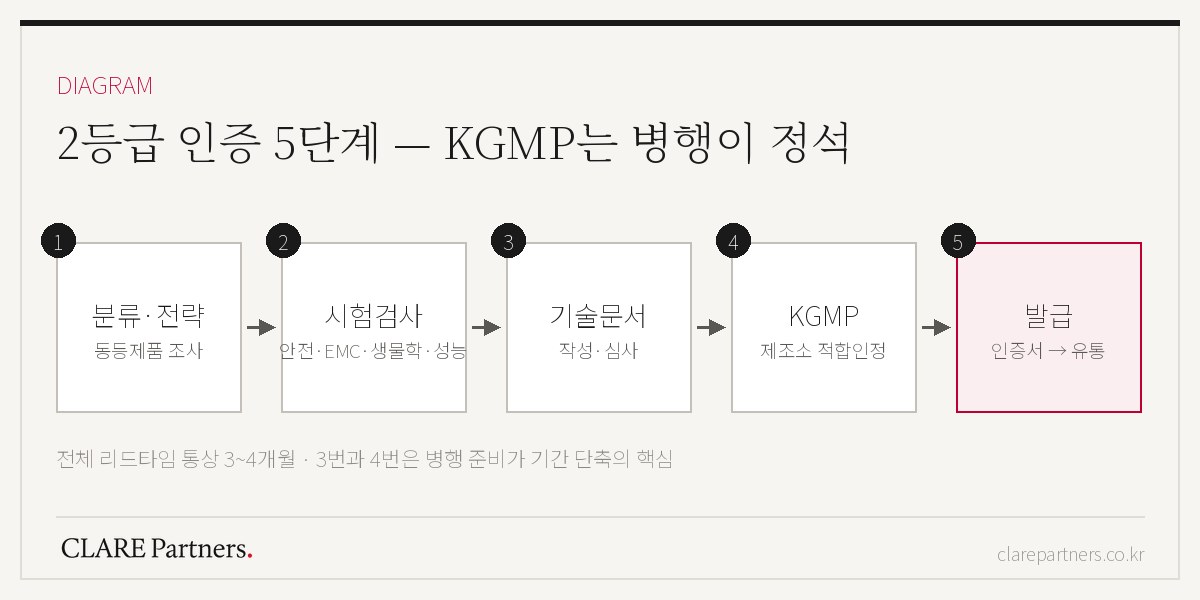
认证流程5步骤
第1步 — 品目分类与战略。 确定品目名称·类别,并调查是否存在已获批的同等产品。同等产品的有无会极大影响审评负担和检测设计,跳过这项调查,后续费用就会膨胀。
第2步 — 检测检验。 在公认检测机构对产品进行检测。基本框架如下。
| 检测 | 对象 | 标准系列 |
|---|---|---|
| 电气·机械安全 | 用电器械 | IEC 60601-1 |
| 电磁兼容(EMC) | 用电器械 | IEC 60601-1-2 |
| 生物学安全性 | 有人体接触部位的器械 | ISO 10993 |
| 性能检测 | 全部品目 | 各品目基准规格 |
海外检测报告的认可范围因案而异,因此先筛选制造商已有资料是节省费用的起点。
第3步 — 编制技术文件·审评。 提交将预期用途、作用原理、结构·规格、检测结果编织成一条完整逻辑的文件。审评员阅读的不是数据的堆砌,而是这条逻辑。
第4步 — KGMP合格认定。 针对制造场所(进口则为海外制造场所)的质量体系审查。详细准备事项在KGMP指南中另有整理,与技术文件并行推进是缩短周期的关键。
第5步 — 认证证书发放 → 流通。
整体周期通常按3~4个月规划较为现实,并会因以下3大变量而大幅波动。
左右周期的3大变量
① 同等产品调查的完成度。 不做调查就开始,检测项目就会被设计得过多(不必要的费用)或过少(因补正而返工)。
② 制造商资料的配合速度。 这是进口项目延误的头号原因。材质信息、检测资料、GMP文件 — 在合同阶段就明确资料提供义务才是稳妥之策。
③ 首次提交的完成度。 补正不是失败,而是流程的一部分,但总周期取决于首次提交的质量。了解各品目审评要点与不了解之间的差距,正是在这里显现。
认证前检查清单
- 确定品目名称·类别,完成同等产品调查
- 取得制造商已有检测资料清单
- 确定韩国国内追加检测项目·机构·日程
- KGMP准备与技术文件并行启动
- 在与制造商的合同中写入资料提供义务
对2类而言,检测设计就是费用。将品目信息和制造商已有资料清单发送至免费事前评估,我们将为您搭建检测项目和预计日程的框架。
常见问题
- Q. 2类医疗器械认证需要多长时间?
- 取决于检测项目数量、补正次数以及制造商的资料配合速度,通常按3~4个月规划日程较为现实。如果没有同等产品或检测需要从头做起,周期会更长。
- Q. 在海外取得的检测报告(CE等)可以用于韩国认证吗?
- 部分可以被认可,但需要逐案确认检测标准·条件是否与韩国要求一致。先筛选制造商已有资料中可被认可的范围,可以减少重复检测的费用。
- Q. 技术文件审评和KGMP可以同时进行吗?
- 可以,并行准备是缩短周期的关键。如果先提交技术文件、再依次等待KGMP审查,整体日程就会相应拉长。