指南按类别2026.07.08
1类医疗器械备案指南
1类医疗器械无需审评,以'备案'方式办理。本文整理了备案流程4个步骤、所需材料,以及实务中最常被驳回的3个环节。
核心摘要 — 1类是四个类别中唯一无需审评、以'备案'即可完成的类别。因此速度快,但也容易掉以轻心。大多数失败并非发生在备案本身,而是发生在其前(品目分类)与其后(标签标识·GMP例外确认)。
什么是1类 — 不是'许可',而是'备案'
医疗器械按潜在风险程度分为1~4类。1类是几乎没有风险的器械,典型例子包括诊疗台、手动轮椅、医用刀剪类等。
该类别在程序上的正式名称是品目备案。无需经过韩国食药处(MFDS)的技术文件审评,通过医疗器械电子民愿窗口办理备案即可,因此是四个类别中最快、成本最低的。不过,如果以"反正只是备案,走个形式"的心态对待,就会遭遇驳回。因为只是没有审评环节而已,各项要求必须全部满足,备案之后的义务也照样存在。
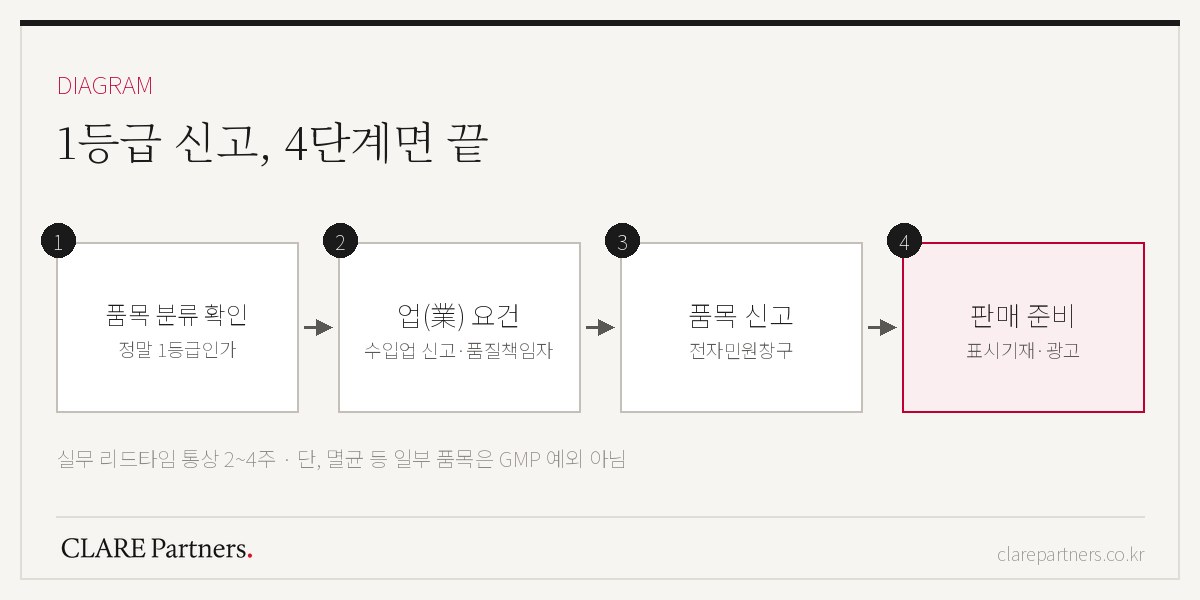
备案流程4步骤
第1步 — 确认品目分类。 首先确定自己的产品是否真的属于1类。名称相似的品目常常分属不同类别,如果这一步出错,之后的所有工作都要重来。(如果对类别没有把握,请先参阅医疗器械注册流程全解中的类别判断标准。)
第2步 — 具备经营资质。 如果是进口产品,在品目备案之前必须先完成进口业备案,其中包括指定质量负责人。这一步的准备事项在进口流程指南中另有整理。
第3步 — 品目备案。 在电子民愿窗口填写并提交备案申请。受理·处理较快,从材料准备到完成的实务周期通常为2~4周。
第4步 — 销售准备。 完善韩文标签标识、如需投放广告则办理事前确认等,满足流通环节的各项要求。
所需材料
| 类别 | 材料 | 备注 |
|---|---|---|
| 产品 | 规格书、结构·原材料资料 | 预期用途的记载是类别判断的依据 |
| 制造商(进口时) | 制造商确认资料、合同关系文件 | 签约前确认能否取得相关资料 |
| 标识 | 韩文标签草案 | 通关·流通环节被指出问题的头号原因 |
| 经营备案 | 质量负责人资格文件等 | 与进口业备案同步办理 |
最常被驳回的3个环节
① 分类判断失误。 "以为是1类,结果是2类"是最常见的事故。根据是否灭菌、有无测量功能、与人体接触方式的不同,类别可能上调。这种情况下需要办理的不是备案,而是从头走2类认证流程。
② 标签标识不完备。 忽略韩文标识要求就先把货物运进来,结果在通关·流通环节被卡住。标签容易因"不是备案材料"而被搁置,但实际业务的瓶颈恰恰产生于此。
③ GMP例外判断失误。 1类的相当一部分被排除在KGMP合格认定对象之外,但灭菌产品等部分品目属于例外。将"1类 = 免GMP"一概而论是危险的。
备案前检查清单
- 取得品目分类(品目名称·类别)的判断依据
- 完成进口业备案(进口时)
- 确认能否从制造商取得所需资料
- 编制韩文标签标识草案
- 确认自己的品目是否属于GMP例外
如果品目分类模糊,或制造商资料的取得没有把握,在备案前先梳理整体结构是最划算的保险。将品目信息发送至免费事前评估,我们将为您确认类别和准备顺序。
常见问题
- Q. 1类医疗器械也需要取得韩国食药处(MFDS)许可吗?
- 不是许可,而是'品目备案'对象。无需韩国食药处(MFDS)的技术文件审评,通过医疗器械电子民愿窗口办理备案即可;不过,进口业备案等经营资质要求和标签标识义务同样适用。
- Q. 1类备案需要多长时间?
- 备案受理·处理本身较快,包含材料准备在内的实务周期通常为2~4周。如果制造商资料提供延迟,周期会相应拉长。
- Q. 1类不需要GMP(KGMP)吗?
- 相当一部分1类品目被排除在KGMP合格认定对象之外,但灭菌产品等部分品目属于例外。备案前务必确认自己的品目是否属于例外情形。