指南GMP2026.07.08
KGMP合格认定指南 — 审查种类、准备文件、海外制造场所应对
KGMP是2类及以上医疗器械的必经关口。本文整理了各审查种类的差异、质量体系文件的框架,以及海外制造场所审查中项目延误的真正原因。
核心摘要 — KGMP审查的不是"产品",而是"制造产品的体系"。它是2类及以上医疗器械事实上的必经关口;在进口项目中,海外制造场所的配合程度直接等于日程。有ISO 13485固然有利,但不能因此免除。
KGMP审查什么
如果说品目注册看的是"这个产品是否安全·有效",那么KGMP(医疗器械生产及质量管理规范)看的是"这个制造场所能否稳定一致地生产出这样的产品"。审查对象是制造场所的质量体系 — 文件、记录、设备、人员,以及这一切实际运转的现场。
2类及以上品目的生产·进口,事实上必须配套相应制造场所的合格认定(参见各类别程序);1类中灭菌等部分品目也属于例外适用对象。
审查的种类 — 何时接受哪种审查
| 审查 | 时机 | 特点 |
|---|---|---|
| 首次审查 | 该制造场所首次取得合格认定时 | 文件+现场,准备负担最大 |
| 追加审查 | 在已合格的制造场所追加品目群等 | 以变更范围为中心 |
| 变更审查 | 场所地址·关键工序等发生变更时 | 评估变更事项的影响 |
| 定期审查 | 有效期届满时更新 | 确认体系是否持续维持 |
如属进口,此审查针对海外制造场所进行,文件审查与现场审查的组合方式视具体情况而定。
质量体系文件的框架
- 质量手册与程序文件 — 覆盖设计·采购·生产·检验·出厂·投诉处理·纠正预防措施(CAPA)的体系
- 产品标准书系列 — 关于该品目'如何制造、如何检验'的标准
- 记录 — 证明确实按程序文件运行的证据。审查中被翻查最多的不是手册,而是记录
- 验证(Validation) — 对灭菌、特殊工序等无法通过结果验证的工序进行事前验证
- 培训·设备管理 — 人员资格与设备的适格性
已建立ISO 13485体系的制造场所,这一框架的相当部分已经具备。实务工作从"已有的"与"韩国要求的"之间的差距分析开始。
海外制造场所审查 — 延误的真正原因
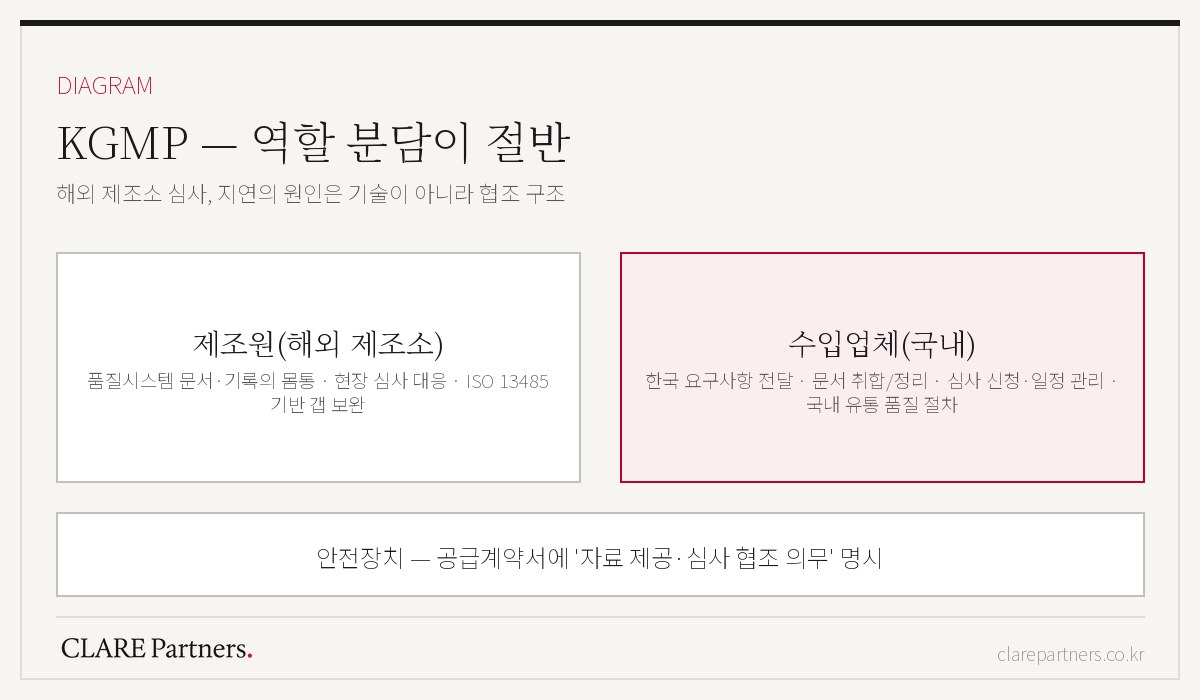
进口项目中KGMP延误的原因,大多不在技术,而在配合结构。
"凭什么是我们?"的壁垒。 站在制造商的立场,KGMP是别国的审查,很容易被排在优先级之后。合同中的审查配合义务条款是唯一可靠的杠杆。
文件的语言与格式。 把制造商的文件整理成韩国审查方能够读懂的形式,这项工作量一旦被低估,日程就会崩塌。
现场准备。 审查当天的提问指向的不是文件,而是现场记录。未经模拟检查就迎接现场审查,细小的记录空白就会演变为补正。
准备检查清单
- 确定目标制造场所·品目群,确认审查种类
- 确认制造商ISO 13485等现有体系 → 差距分析
- 在合同中写入资料提供·现场审查配合义务
- 预留文件汇总·翻译·整理的日程
- 制定现场模拟检查计划
- 设计与技术文件审评并行的日程
KGMP的一半在于与制造商的协作设计。将制造场所信息(国家、现有认证、品目群)发送至免费事前评估,我们将为您起草审查种类和准备范围的差距分析初稿。
常见问题
- Q. KGMP必须在什么时候之前取得?
- KGMP与品目认证·许可相互衔接,产品要出厂·销售,相应制造场所必须已取得合格认定。与技术文件审评并行准备,是缩短整体周期的正规做法。
- Q. 海外制造商已有ISO 13485认证,可以免除KGMP吗?
- 不能免除。ISO 13485是准备KGMP的良好基础,但KGMP是依据韩国要求进行的独立合格认定程序。不过,13485体系完善的制造场所,其文件差距分析和补充范围会大幅缩小。
- Q. 我们作为进口商要准备的事项和制造商要准备的事项如何划分?
- 质量体系文件和现场审查应对的主体是制造商,进口商则负责传达韩国要求、汇总文件、申请审查与日程管理,以及韩国国内流通环节的质量程序。如果不在合同中明确这一角色分工,项目就会卡在中间。